
Pathogenesis driven by RNAs with expansion of trinucleotide repeats: mechanisms and therapeutic strategies
Okres realizacji: 1.10.2018 – 29.06.2022
Wartość projektu: 3 499 222,00 PLN
Wkład Funduszy unijnych: 3 499 222,00 PLN
1. BACKGROUND AND OBJECTIVES
More than 20 human inherited diseases are caused by expansions of simple repeat sequences, mostly trinucleotide repeats. Among those typically multisystem ailments are fragile X syndrome (FXS, expansion of CGG repeats), Huntington’s disease (HD; expansion of CAG repeats), myotonic dystrophies (DM; expansion of CTG repeats) and Fragile X-associated tremor/ataxia syndrome (FXTAS, expansion of CGG repeats). Depending on location of the triplet repeat expansion within a gene, their pathogenic effect can be visible on transcriptional level (e.g. FXS; loss of gene function) and post-transcriptional level either by gain of mutant RNA function (e.g. DM and FXTAS) or gain of mutant protein function (e.g. HD but also FXTAS). Often the expansions are located within genomic regions that are transcribed but not translated, giving rise to RNA dominance, a process in which transcripts from the mutant allele have a direct deleterious effect. On the other hand, untypical translation of toxic proteins having long mono-amino acid tracts from normally untranslated regions of mRNAs is another feature of majority of these diseases [1].
1.1. DM1 is caused by insufficiency of MBNL protein activity due to their sequestration on toxic CUGexp RNAs 1.1.1. With a worldwide incidence of 1 out of 8,000, Myotonic Dystrophies (DM) are the most common forms of adultonset muscular dystrophy [2]. Myotonic Dystrophic patients are characterized by multiple symptoms including muscle atrophy, myotonia, cardiac conduction defects as well as cataracts, cognitive impairment and endocrine disorders. Life expectancy of these patients is greatly reduced, reflecting an increase in death due to respiratory diseases caused by the muscle atrophy. There are two forms of the disease, DM1 and DM2. DM1 is an RNA dominant disorder caused by expansion of a CTG repeat in the 3’-UTR of the DM protein kinase (DMPK) gene. Individuals with classical DM1, typically inherit gene alleles with hundreds of CTG repeats. However, the mutation is quite unstable in somatic cells and it continues to expand during postnatal life, reaching lengths of several thousand repeats in muscle and cardiac tissue. The DMPK transcripts containing these highly expanded CUG repeats (CUGexp) are subjected to splicing but are retained in the nucleus in discrete foci. Their nuclear retention is partly a function of the interaction of CUGexp RNA with poly(CUG) binding proteins, such as, splicing factors in the Muscleblind-like (MBNL) family [3]. The pathogenic effects of CUGexp RNA are due in part to sequestration of MBNLs, which results in misregulated alternative splicing (Fig. 1) or decay of transcripts that these proteins normally regulate. Another pathogenic effect of the mutant DMPK mRNA is to induce post-translational upregulation of another RNA binding protein, CUGBP1. According to unknown mechanism this protein is abnormally phosphorylated and predominantly localized in nuclei of DM1 cells.
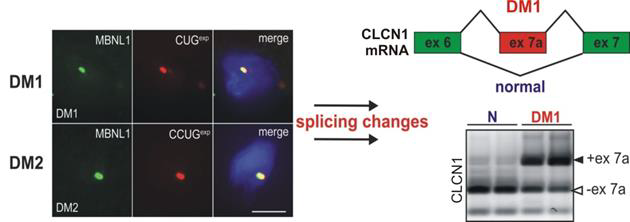
Both, sequestration of MBNLs in nuclear CUGexp foci which diminishing their functional cellular levels and upregulation of CUGBP1 strongly influence splicing abnormalities of several transcripts in DM muscles and neurons. Both proteins act as antagonistic splicing factors and control developmentally regulated pre-mRNA maturation and their functional imbalance leads to embryonic pattern of splicing in postnatal development in DM1 tissues [3, 4].
1.1.2. The family of muscleblind-like proteins (MBNL1, 2 and 3) belongs to tissue-specific regulators of RNA metabolism [14]. The primary role of MBNLs is regulation of alternative splicing in pre-mRNA targets, which includes splicing of hundreds of alternative exons and polyadenylation site selection [18-20]. MBNLs are also implicated, to a lesser extent, in regulation of subcellular localization of particular mRNAs [19, 21], mRNAs stability [18, 19, 22] and micro-RNA processing [23]. Out of the three paralogs, MBNL1 serves a primary splicing function in the skeletal and cardiac muscles [14]. MBNL2 is highly expressed in the brain [24]. MBNL3 is mostly present at early stages of tissue development [25, 26]. MBNL paralogs, recognize RNA targets by four zinc fingers (ZFs) serving as RNA binding domain [27]. The ZFs are organized in two tandems (ZF1-2 and ZF3-4) [27, 28] divided by an elastic and unstructured linker which may facilitate MBNL proteins to bind to remote RNA sequence motifs [29, 30]. MBNL1 fragment encompassing ZF1-2 and a linker is the smallest entity required for splicing regulation [31-33]. Various analyses including conducted by us whole-transcriptome CLIP-seq indicated a minimal RNA sequence motif recognized by MBNLs in RNA targets which is 5ʹ-YGCY-3ʹ (where Y is a pyrimidine) [17, 19, 22, 24, 34]. MBNL paralogs are represented by several splicing isoforms differing in a presence of residues encoded by alternative exons. These amino acid sequences determine MBNL binding affinity (exon 1 and 3 encoding residues), cellular localization of the protein and its ability to form dimers (exon 5 and 7) [35-37]. Recently performed NMR spectroscopy analyses, as well as other biochemical and high-throughput studies demonstrate that RNA-binding domain of MBNL1 binds both single-stranded RNAs and RNAs distinguished by thermodynamically semistable secondary structure undergoing partial unwinding upon protein binding [29, 30, 38, 39]. The structure of MBNL, RNA-binding domain only or a full length protein, associated with RNA has not been determined yet. Thus, it would be compelling to gain deeper insight in conformational changes of protein domains implicated in RNA binding and proteinprotein interactions upon binding of various RNAs and how these changes contribute to mechanism of splicing driven by MBNL (Objective 1 of this project). MBNL binding to particular regions within pre-mRNA targets is linked with the protein splicing function. MBNL binding within regulated alternative exons or sequences of upstream introns results in alternative exon skipping from the mature mRNA [19, 24] while binding within introns located downstream of the alternative exon promotes alternative exons inclusion [19, 24]. While binding within downstream introns, MBNL perhaps affects spliceosome formation by enhancement of binding of U2 auxiliary factor 65 kDa subunit (U2AF65) responsible for 3ʹ splice site definition [40]. In contrast, when MBNL binds within upstream introns the protein plays a role of U2AF65 competitor probably by introducing RNA structural changes upon binding leading to impeded U2AF65 association with RNA [41]. Moreover, MBNL associated with RNA affects function of other trans-acting splicing factors including CELF1 (CUG-binding protein elav-like family member 1) [42], RBFOX (RNA-binding protein, fox-1 homolog) [43], DDX helicases which induces local unwinding of the RNA secondary structure [44] and described recently by us SRSF1 (serine/arginine-rich splicing factor 1) [45]. Hence, the mechanism of alternative splicing regulation by MBNL is still unclear. The question arises whether MBNL constitutes only a steric hindrance preventing splicing enhancers from binding leading to alternative exon exclusion. It is also unknown by what means, considering also other protein mediators, MBNL couples with spliceosome machinery leading to alternative exon inclusion into mRNA (Objective 2 of this project). The MBNL paralogs specifically and dynamically interact with CUGexp repeats in DM1 cells [17, 27] which is underlain by a high MBNL-binding affinity for the repeats emerged in biochemical assays [17]. The MBNL paralogs also vary in mobility in nuclear foci. MBNL3 is distinguished by the slowest rate of binding and dissociation from CUGexp repeats [17]. The progression of somatic expansion leads to enhanced MBNL sequestration and constrains MBNL mobility to intra-foci binding sites on CUGexp transcript [17]. However, the precise mechanism and kinetics of this pathogenic CUGexp/protein interaction is unknown and question arise why the sequestration process of MBNLs is very efficient (Objective 1 of the project). DM-like features were reproduced in Human Skeletal Actin-Long Repeat (HSALR) mouse which show muscle-specific expression of the human alpha (skeletal) actin transcript (HSA) that contains ~ 250 CUG repeats in the 3′-UTR [5]. The CUGexp transcripts are retained in the nucleus, they sequester MBNL proteins, and they induce splicing changes and repetitive action potentials (myotonic discharges) similar to patients with DM1. DM-specific aberrant splicing pattern was reproduced also in other mouse models such as Mbnl1 knockout mouse (Mbnl1ΔE3/ΔE3) and in mice overexpressing CUGBP1 protein [6, 7]. Mbnl1ΔE3/ΔE3 mice showed for example aberrant splicing of CLCN1 and myotonia, abnormal muscle histology and cataracts similar to what is found in DM1 patients. Therefore, overall reduction of the MBNL1 mimics the effect of the DM1 mutation. To date, four main strategies of targeting expanded repeat RNA in experimental therapy of DM were described: (1) DNA editing in DMPK loci containing CTGexp to remove this pathogenic sequence from mutated gene [8]; (2) degradation of mutant DMPK transcript using RNA interference (RNAi) tools [9] or antisense oligonucleotides [10, 11], (3) inhibition of pathogenic interaction of repeat RNA with nuclear proteins (e.g. MBNLs) by either antisense oligomers complementary to CUG repeats [12, 13] or CUG repeat hairpin-specific small compounds [14, 15], or (4) overexpression of MBNLs using gene therapy tools [16]. Exogenous overexpression of MBNL1 in vivo rescued global splicing abnormalities and muscle myotonia in the HSALR mouse model, what was correlated with restitution of splicing pattern of CLCN1 and correction of splicing profile of several other pre-mRNAs [16]. However, the histology of muscle showed no restoration of myofiber structure, which may be due to insufficient or not equal overexpression of MBNL1. Moreover, the isoform of MBNL1 used for studies might have been inefficient as our recent analysis reports of RNA-substrate dependent distinct activities of MBNL paralogs and their isoforms [17]. In contrast, the overactivity of exogenous MBNL1 in cells leading to deleterious effects may also occur due to the overexpression of an entirely active protein. Therefore, there is a necessity to design an MBNL-like derivatives exerting a minimal effect on natural RNA targets but with high binding affinity for CUGexp as well as a gene therapy tool prepared in a way to introduce the protein into cells and regulate its cellular level (Objective 3 of the project).
1.2. FXTAS is caused by both toxic CGGexp RNA and products of untypical translation of this RNA
1.2.1. Fragile X-associated tremor/ataxia syndrome (FXTAS) is an X-linked inherited late onset neurodegenerative disorder causing tremor, ataxia, brain atrophy, cognitive loss, dementia and early death in some individuals. The disorder is caused by an expansion of 55-200 CGG repeats in the 5’-UTR of the FMR1 gene encoding FMRP, also named premutation (PM) [46]. The prevalence of PM carriers in the general population is high, with estimates of 1:250 for females and 1:400 for males [47]. The chances of developing FXTAS increase dramatically with age, with approximately 30% of male and 8-11% of female PM carriers over the age of 50. Altogether, the incomplete penetrance results in estimates of 1:~3,000 males who are at risk for developing FXTAS [48]. Females may also develop FXTAS, although less commonly compared to males, presumably because of the protective effect of the second X-chromosome.
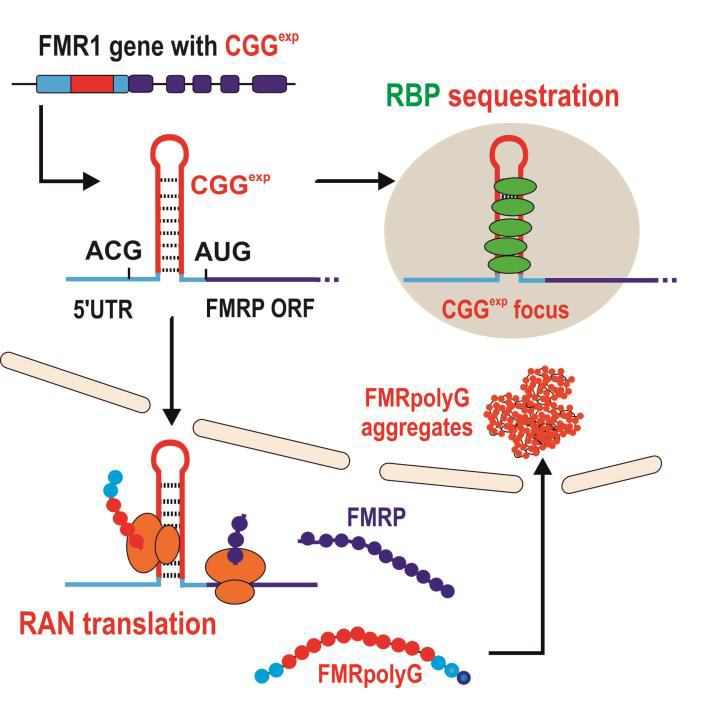
The hallmark of FXTAS neuropathology (in postmortem brain tissue) is the presence of ubiquitin-positive intranuclear aggregates in both neurons and astrocytes that also contain mutant FMR1 mRNA [49]. Also, PM carriers with and without FXTAS show up to 8 fold elevations in FMR1 mRNA in leucocytes and 2-3 fold elevations in brain tissue due to increased FMR1 transcription and paradoxically slightly reduced levels of FMRP protein expression [50]. These observations have led to the proposal that FXTAS results from a toxic RNA gain-of-function, in which mutant RNA containing expanded CGG repeats (CGGexp) would be pathogenic by sequestrating specific RNA binding proteins within nuclear aggregates [51], as was described above for DM1. This would result in loss of their normal cellular function and ultimately in neuronal dysfunction and cell death. Indeed, it was identified that Sam68 and other RNA binding protein DGCR8 and its partner, DROSHA, are sequestered within CGGexp RNA aggregates (Fig. 2) [52, 53]. Sam68 is involved in the alternative splicing of several mRNAs implicated in normal neurogenesis. Sequestration of Sam68 results in abnormal splicing of target mRNAs. DROSHA-DGCR8 is the enzymatic complex that participates in processing microRNAs. Consequently, the level of mature microRNAs is reduced in brain samples of patients with FXTAS, resulting in neuronal cell degeneration. Importantly, the sole over-expression of DGCR8 rescues the neuronal cell death induced by expression of expanded CGG repeats in primary cultures of murine neurons. Thus, the sequestration of DGCR8 is a leading event in the neuronal degeneration observed in FXTAS.
1.2.2. The mechanism of RAN translation is as yet unknown.
Recently, an additional mechanism of toxicity that is triggered by CGG repeats associated non-AUG initiated (RAN) translation has been proposed to underlie pathology in FXTAS [54-56]. This is based on evidence that trinucleotide repeats, even if not present in coding sequence, can be translated into protein even if they do not reside in an AUG-initiated open reading frame (ORF), and such translation can occur in all three possible ORF’s of a transcript generating multiple potentially toxic products from a single repeat tract. In the case of FXTAS it has been proposed that RAN translation initiated in the 5’UTR of FMR1 mRNA results in the production of a cytotoxic polyglycine- and polyalanine-containing protein, called FMRpolyG and FMRpolyA, respectively. Indeed, the presence of both FMRpolyG and FMRpolyA was demonstrated in brain tissue from patients with FXTAS [56, 57]. Moreover, recently published data indicate that probably not protein sequestration on RNA with CGGexp but mainly CGG repeat associated non-AUG initiated (RAN) translation can underlie FXTAS pathogenesis. Mice expressing full length human FMR1 5’UTR with CGGexp showed presence of FMRpolyG aggregates and locomotor deficiency in contrast to mice expressing CGGexp outside of FMR1 sequence context. Furthermore, presence of sequence flanking CGG repeats in FMR1 5’UTR enables transport of transcript to cytoplasm and makes it available for translation, whereas its absence increases CGGexp RNA intranuclear accumulation. Another reason for the importance of RAN translation in FXTAS is connection between FMRpolyG aggregation and toxicity-causing architectural changes of nuclei in neural cells. FMRpolyG sequesters lamina-associated polypeptide beta (LAP2β), which is responsible for lamina B1 and B2 organization near nuclear membrane [56]. Although new data regarding the RAN translation mechanism in FXTAS are still emerging, many aspects of this process remain unexplained. It is known that biosynthesis of RAN translation toxic proteins is 5’-cap and elF-dependent. Besides, potential RAN translation initiation ACG codon (and in some extend other triplets) was identified in FMRpolyG reading frame. Replacing this codon with canonical AUG codon significantly enhanced FMRpolyG production. Efficiency of CGG repeats RAN translation is also repeat-length and reading frame dependent. Polyglycine stretch is responsible for FMRpolyG aggregation which increases with increasing repeat size [54, 55, 58]. It was also shown that interaction of RNA binding proteins TDP-43 and hnRNPA2/B1 reduced CGG associated neurotoxicity in Drosophila FXTAS model [59]. We believe that questions concerning importance of other sequence and RNA structural determinants located in 5’UTR of FMR1 in initiation of RAN translation and their efficiency should be addressed. Extremely important is also to identify other transfactors taking part in RAN-dependent biosynthesis and nuclear aggregation of FMRpolyG and degradation of this protein (Objective 4 of this project). Important insight into molecular pathomechnism of FXTAS has been given by mouse model studies. Currently there are a few most relevant FXTAS models. Knock-in models (so called Dutch mouse and NIH mouse) possess expanded CGG repeats with or without human flanking regions in mouse Fmr1 sequence. FXTAS features observed in both models include elevated Fmr1 mRNA expression, decreased level of FRMP, ubiquitin-positive intranuclear inclusions, FMRpolyG expression (only Dutch mouse) and some neurobehavioral symptoms [60-63]. Recently doxycycline-inducible FXTAS mouse model with expression of 90 CGG repeats outside of the context of Fmr1 gene was generated. It enables controlled activation and stopping of transgene expression. Inducible mouse model show presence of intranuclear ubiquitin-positive inclusions, FMRpolyG expression and FXTAS neurobehavioral features [64]. Appearance of motor impairments and emotional disturbance makes this model the most applicable to test potential therapeutic strategies [65]. So far only a few papers addressed a question regarding targeting of mutant FMR1 as potential therapeutic strategy in FXTAS. Recently, Disney’s lab demonstrated capacity of a few small molecules which can specifically bind to RNA CGG repeats and induce correction of missplicing of many mRNAs caused by displacement of SAM68 splicing factor from pathogenic sequestration on CGGexp. Moreover, some of these compounds significantly reduce CGG RAN translation but not canonical translation from FMRP ORF in FXTAS cell models [66-68]. Better understanding of molecular pathomechanism of FXTAS give a chance for elaborating other causative treatment strategies. Experiments with FXTAS inducible mouse model show that stopping and to some extent reversing of disease progression is possible. For this reason therapeutic strategy using other small compounds targeting CGGexp but also antisense oligonucleotides either blocking translation of FMRpolyG and FMRpolyA or inducing mutant transcript degradation seems to be promising approaches (Objective 5 and 6 of this project). However, eventual consequences on FMRP loss resulting from this therapy should be taken into consideration [58].
2. METHODOLOGY
2.1. Mechanism of MBNL binding to RNAs and MBNL-like therapeutic peptides binding to CUGexp
2.1.1. Objective 1: The mechanism of RNA recognition by MBNL proteins (the project for post-doc)
MBNL1 specifically recognizes the YGCY motif within RNA primary sequence, although the presence of the RNA motif is not the only determinant of high binding affinity or splicing activity of the protein. Our preliminary studies indicated a superior role of YGCY motifs’ arrangement as well as the position of RNA motifs on the RNA structure over the number of YGCYs in splicing regulation. Contiguous four YGCY motifs, but not interspaced, were favored by MBNL splicing activity but not RNA binding. Intriguingly, RNA regulatory elements with certain composition of four YGCY motifs were effectively bound by MBNL1, although served as weak cis-acting elements in splicing regulation (Fig. 3a,b). Therefore, we want to address a question, why RNA binding affinity not correlate well with splicing activity of MBNLs?
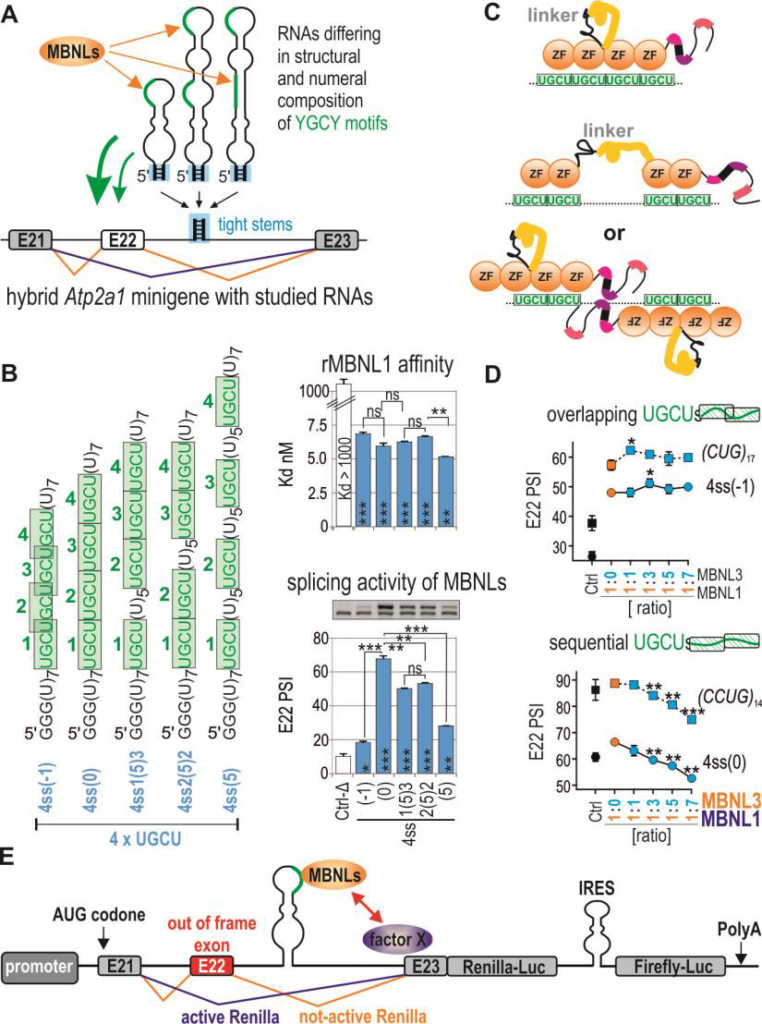
First we will test several other MBNL-binding sequences with different YGCY-motifs arrangement and located either in intron (splicing activation) or exon (splicing repression) of hybrid minigene construct which we previously developed [44]. Next, we want to test several possible hypotheses to answer this intriguing question: (1) MBNLs bind four YGCYs using four ZnFs and the difference in structure of targeted RNA impact MBNL protein conformational changes what affects splicing activity, (2) only two ZnFs are engaged in YGCYs recognition and depending on arrangement of these motifs either one or two MBNLs can bind, but interplay between two RNA associated MBNLs may induce stronger effect on splicing, (3) MBNL binds distinct RNAs with the same affinity but different kinetics (Fig. 3c). To verifiy these hypotheses we want to use few experimental settings (approaches?). First, to analyze the differences in global structure of RNA/protein complexes the Small Angle X-ray Scattering (SAXS) experiments will be done with different MBNL isoforms [16] and few model RNA substrates differing in the number and arrangement of YGCYs. SAXS is biophysical technique that is useful for the study of the structure, conformational changes and dynamics of the macromolecules in solution. Moreover, to study the size of RNA/protein complexes and the number of MBNLs associated with single RNA we will utilize the Multi-angle Laser Light Scattering (MALLS) combined with size exclusion chromatography (SEC). Secondly, in collaboration with Dr. Zawadzki from our University we are going to adopt single molecule microscopy to study the nature of interaction between different fluorescently tagged MBNLs and different RNA molecules (Dr. Zawadzki has years of experience in single-molecule studies [69-77]. We will perform the tracking of labelled MBNLs inside living cells and also perform in vitro single-molecule analysis of MBNLs present in cellular extracts (here specific RNA is attached to the surface) to study the kinetics and nature of RNA/protein interaction (we want to analyze in the same time pairs of overexpressed MBNLs with different labels). Using this unusual single-molecule platform we are also going to answer other important question, how single MBNL protein binds single CUGexp RNA? To better understand the mechanism of MBNL sequestration in DM we are going to study potential interactions between MBNLs on the same RNA to answer whether binding of different MBNLs is either independent or synergistic. Other experiments will be focused on kinetics of dissociation and association and dynamics of replacement of different MBNL paralogs and their isoforms on CGGexp (see also 2.1.3). In our preliminary experiments we also found that splicing activity of different MBNLs would be similar or completely different depending on a structure of targeted RNAs (Fig. 3d). We are going to study this phenomenon using the battery of assays described above and assays allowing detection of potential protein dimerization on RNA target (co-immunoprecipitation and enzymatic complementation assays for MBNL pairs fused with different GFP or luciferase fragments).
2.1.2. Objective 2: MBNL-associated protein interaction networks (the project for post-doc with support of other team members)
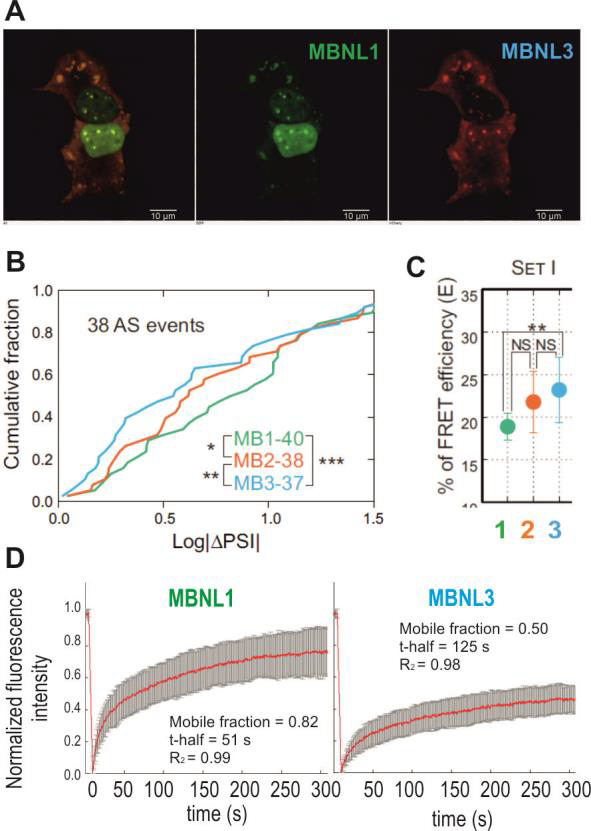
To regulate alternative splicing MBNL proteins bind to specific sequences/structures in pre-mRNAs and after subsequent interaction with other trans-factors impact on exon definition and spliceosome assembly. The aim of this part of the project is to find factors involved directly or indirectly in MBNL-dependent splicing using whole genome-based approaches, such as CRISPR/Cas9 or RNAi screens for essential genes. In this approach the individual gene or pool of genes will be knockedout/ silenced in engineered cell model and the final effect on MBNL-dependent splicing will be measured in high-throughput scale. As the splicing is co-transcriptional process it would be regulated by several protein families including not only RNA binding proteins (e.g. splicing factors, splicing enhancers or silencers, RNA helicases involved in static splicing mechanism) but also chromatin binding proteins (e.g. Pol II-associated proteins; involved in kinetic splicing mechanism which depends on the speed of transcription). We are going to test at least two different focused gRNA/siRNA libraries covering these two gene families. More recently, a set of 1155 siRNAs targeting 375 different genes predicted or known to encode regulators or components of chromatin was used to study interactome of U2 snRNP [78]. Now, CRISPR technology seems to be less affected by time-dependent genetic drift and by off-target gRNA cytotoxicity compared to RNAi screening platform. For that purpose we are going to prepare two high-throughput cellular screens with stable and inducible expression of reporter Atp2a1 minigene with MBNL-dependent exon 22 (E22) and sequences encoding two luciferases. This reporter translates transcriptional activity into Renilla-luciferase luminescence (normalization control), while inclusion of E22 allows for in-frame splicing of the Firefly mRNA and thereby produces Firefly-luciferase activity (Fig. 3e). This reporter and its variant missing the MBNL-binding sequence (negative control) will be inserted into the genome of WT MEFs or MEFs from Mbnl1, 2 double knock-out mouse (lack of MBNLs). After identification of positive screening hits each candidate gene will be validated using other MBNL-sensitive minigenes and MBNL-sensitive exons from endogene transcripts to identify partners crucial in MBNL network (potential direct and indirect interaction partners). Further study will identify proteins involved in alternative splicing regulation via direct interplay with MBNLs. Such interaction will be confirmed in pool-down experiments or yeast two-hybrid system for different MBNL paralogs and their splicing isoforms. Domains of MBNL important for this interaction will be defined by mutagenesis. We expect that among hits from screening we also may identify factors which impact expression of either MBNLs on transcriptional or post-transcriptional level. This knowledge may have translational potential to find agent which can increase steady state level of MBNLs in therapy of DM. 2.1.3. Objective 3: Engineered MBNL-like proteins as potential therapeutic agent in DM (project for post-doc) The goal of this part of the project is to develop MBNL-like molecule with extreme ability to bind selectively CUGexp to use it as efficient competitor of endogenous MBNL sequestration in DM. Our preliminary results obtained based on an MBNLsensitive minigene with a replaceable MBNL-binding cassette (Fig. 3a) [45], shed more light into understanding of various binding affinity and splicing activity of MBNL paralogs and isoforms depending on recognized RNAs. MBNL-mediated splicing profile of alternative exons was considerably affected by the arrangement of YGCY motifs within RNA regulatory elements (Fig. 3b) and MBNL paralog type (Fig. 3d, 4b). For example, four CUG repeats-containing RNA cassette was hardly responsive to MBNL1-mediated splicing regulation but binding affinity is high. Due to these promising findings combined with observations of longer retention of MBNL3 paralog on CUGexp RNA in nuclear foci in DM1 cells (Fig. 4a,c,d) [17], we are going to design MBNL3-like derivative molecule distinguished by high affinity for the toxic RNA repeats and by low splicing activity. Because the linker sequence dividing ZF tandems most likely allows MBNL to bind to distant motifs, in our approach we will remove whole or the part of the linker sequence from the protein. MBNLs most likely interact with other trans-acting factors in splicing regulation; therefore, we will aim at removal of protein regions involved in protein-protein interactions. In order to enhance the available concentration of MBNL3-derivative molecule in nuclei, the protein will be also supplemented with a domain encoding a nuclear localization signal (NLS). Other important property of the MBNL3- derivative molecule would be stabilization of MBNL3-like/CUGexp complexes. To achieve that goal we plan to incorporate into MBNL3 a sequence triggering multimerization, which, to a great extent, may be derived from other proteins. One example is PsbF transmembrane helix dimer containing six amino acids allowing dimerization and stability of proteinprotein complexes [79]. All designed MBNL3-like constructs in fusion with fluorescent tag first will be tested in cells with and without CUGexp. We will study (i) mobile fraction of MBNLs in nuclear CUGexp foci using FRAP and FRET assays [17], (ii) kinetics of dissociation of designed MBNLs with CUG repeat hairpin using single molecule microscopy and (iii) alternative splicing changes of MBNL-dependent exons (RT-PCR) to assess splicing activity of studied construct in WT cells and measure the rescue of endogenous MBNLs from sequestration in DM cells. Constructs showing low or no splicing activity and efficiently interacting with CUGexp will be used to generate AAV-based vectors for in vivo experiments (they will be carried out by a specialized company for such construct preparation).
Moreover, our ambition is to overexpress the MBNL3-like transgene in the way to strictly control its cellular concentration. Our previous study showed that the level of MBNLs in cells undergo autoregulation feedback loop [80]. Therefore, we want to put MBNL3-like ORF under control of MBNL level-sensitive minigene unit as shown in Fig. 3e (luciferase coding cassette will be replaced with MBNL coding cassette). In DM1 cells the expression of MBNL3-like oligopeptide should be significantly reduced when the level of available endogenous MBNLs rescued from sequestration on CUGexp will be sufficient, because the out of frame E22 will be included into transgene mRNA. The AAV-based MBNL3-like constructs will be first tested in different cellular models. Then, they will be delivered into HSALR, the best mouse model of DM1, in Dr. Swanson lab (partner in this project). After three weeks from viruses injection the functional test will be done (e.g. electromyography to study myotonia) and molecular examination of muscle samples will be performed (e.g. immunostaining of MBNL3-like-myc-tagged and endogenous MBNL1; splicing analyses of MBNL-dependent exons using RT-PCR), as we previously described [9, 12, 13].
2.2. Mechanism of RAN translation from CGGexp containing RNA and inhibition of this process
2.2.1. Objective 4: Determinants affecting RAN translation from FMR1 mRNA carrying CGGexp permutation (the project for first PhD student)
One of pathogenic agent in FXTAS are FMRpolyG and FMRpolyA proteins (either soluble or aggregated form) which biosynthesis proceed through untypical RAN translation from mutant FMR1 mRNA containing expanded CGG repeats (Fig. 2). Probably the main factor which drive efficiency of RAN translation is formation of ribosome initiation complex on the near cognate start codon present in 5’UTR. This phenomenon may depends on at least three factors: (i) nucleotide sequence in vicinity of this codon, (ii) stable secondary and tertiary structure of RNA formed by the sequence located downstream of this codon and (iii) binding of some specific proteins in 5’UTR sequence/structure. All of them may affect the speed/kinetics of ribosome scanning or ribosome pausing. We are going to check all these possibilities using modified genetic construct encoding FMRpolyG/A fused with luciferase or GFP (Fig. 5a) . Primary and secondary RNA structure elements of 5’UTR of FMR1 engaged in RAN translation. Firstly, we are going to experimentally determine the RNA structure formed in solution by 5’UTR of FMR1 using a battery of enzymatic and chemical RNA structure probing reagents, as we previously described for many RNAs [80, 82, 83]. Based on this structure we will introduce several mutations to this RNA to affect the changes in different structural motifs (stabilization or destabilization of selected hairpin structures which may affect ribosome pausing) or in the sequence close to near cognate start codons for RAN translation. Then, we will be testing the effect of these mutations on efficiency of FMRpolyG/A production in cellule to define structures of studied 5’UTR crucial for efficient RAN translation. In the same time, we want to perform ribosome profiling experiments (RNA deep sequencing of ribosome associated RNAs) for endogenous (cells derived from patients and healthy subjects) and exogenously introduced into cells mRNAs of FMR1 containing mutations described above and different length of CGG repeats Finally, we want to confirm ribosome pausing in 5’UTR of FMR1 and characterize the structural signature of cic-elements which either enhance or decrease the efficiency of RAN translation [81].
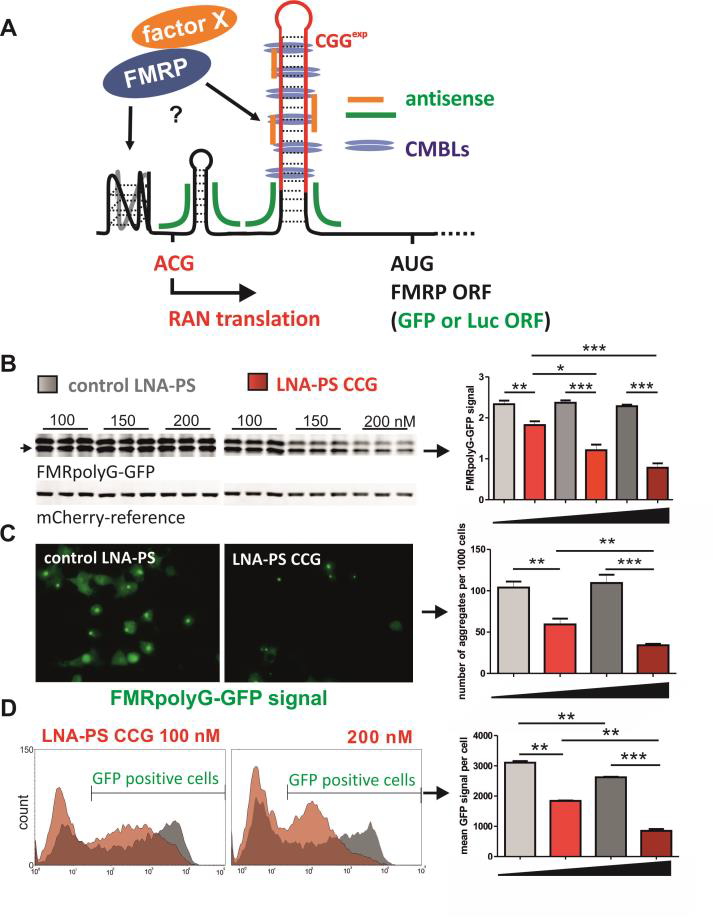
Potential impact of FMRP on its biosynthesis and RAN translation. FMRP is a product of normal translation from FMR1 mRNA. It is RNA binding protein highly expressed in brain, where it is responsible for synapse maturation and plasticity. Among other functions like mRNA transport and splicing, FMRP is considered to be translational repressor [84, 85]. The RNA-binding domain of FMRP has high affinity to G-rich sequences which form RNA Gquadraplexes. Such structure was identified in 3’-part of FMR1 mRNA what is consistent with the fact that FMRP binds its own transcript. It was shown that this interaction do not influence FMR1 mRNA translation but is responsible for its alternative splicing [86-88]. Previously we and others showed that the CGG repeat RNA form hairpin-like secondary structures with some features characteristic for G-quadruplexes [89, 90]. Moreover we found that in 5’UTR of FMR1 close to near-cognate ACG start codon some other sequence motifs has also potential to form such untypical structures. Therefore, we ask whether FMRP can bind to expanded CGG repeats or other sequences within FMR1 5’UTR and influence efficiency of RAN translation? First, we want to perform the in vitro analysis of recombinant rFMRP binding to FMR1 5’UTR. Then, we will investigate the effect of FMRP overexpression or siRNA induced silencing on both FMRpolyG biosynthesis and translation of FMRP using engineered COS-7 cells (with WT or mutant 5’UTRs deprived of potential Gquadruplexes) and patient derived cells. As FMRP is responsible for FMR1 mRNA transport in neurons [85] we want to check whether binding of FMRP to FMR1 5’UTR with normal or expanded CGG repeats affects cellular transport of its mRNA. Neural cells transfected with genetic construct encoding 5’UTR of FMR1 fused with ORF for mCherry will be monitor to visualize fluorescent protein biosynthesis in dendritic spines (FMRpolyG-mCherry). This subtask will be executed in collaboration with Dr. Dziembowska, neurobiologist from University of Warsaw [91]. Other cellular components (trans-factors) engaged in FMRpolyG biosynthesis, aggregation and degradation. In parallel we want to identify trans-factors affecting efficiency of RAN translation from mutant FMR1 mRNA but also define natural factors responsible for degradation of toxic FMRpolyG. To achieve this goal we want to utilize RNAi screening (as described in 2.1.2 for MBNL network) using cell model with stable expression of Renilla-luciferase reporter fused with FMR1 5’UTR with CGGexp in polyglycine reading frame. The control Firefly-luciferase construct will be used to identified general factors affecting translation (both cassettes will be expressed from bidirectional promoter; we have already prepared such construct). We are going to use at least two siRNA libraries representing genes for RNA-binding proteins and proteins which involved in metabolism of other proteins (degradation, aggregation etc.). The secondary screening will be performed using construct having GFP-tagged FMRpolyG and FMRpolyA and microscope-based quantification of total fluorescent signal or signal from aggregates. Finally, the function of individual gene-candidates in RAN translation/aggregation will be analyzed with more details.
2.2.2. Objective 5: RNA structure-based designing of antisense oligonucleotides which block RAN translation (the project for second PhD student)
We are going to develop a gene-targeting approach to test whether antisense strategies targeting mRNAs containing expanded CGG repeats can reduce efficiency of RAN translation. Based on experimentally predicted structure of 5’UTR of FMR1 (2.2.1) we will design antisense blockers which consist of different nucleic acids analogs complementary to sequences flanking CGG repeats which not induce degradation of targeted transcript but reduce initiation of RAN translation from near-cognate start codon. We postulate that the RNA structure in 5’UTR region is important for ribosome pausing what result in efficient initiation of RAN translation. As antisense reagents we would utilize morpholino oligomers, PNAs (peptide nucleic acids), 2’OMe-RNAs, LNA/DNAs (locked nucleic acids) mixmers. These types of antisense molecules interact more effectively with targeted RNA sequences and are much more stable in the cellular environment than natural nucleic acids. Moreover, such molecules do not induce degradation of targeted RNAs. Previously, we used with success all these oligomer types as a blocker of RNA/protein interaction in DM models, but our preliminary results suggest that antisense blockers targeting directly the CGG repeats may also prevent RAN translation of FMRpolyG; however, unfortunately affect also expression of FMRP. In a first step all designed oligomers will be tested in vitro to determine their affinity to FMR1 5’UTR RNA. Then, they will be tested in engineered COS-7 cells, fibroblasts derived from FXTAS patients and neuronal cells isolated from FXTAS mouse brain. These cells are an ideal starting point for testing antisense strategies for efficacy since they rapidly show nuclear FMRpolyG inclusions as well as decreased cell viability in culture. We will use lipofection reagents to deliver antisense blockers and well-established delivery systems for antisense oligomers (liposome/2’OMe, endoporter/morpholino complex or PNA conjugated with (Arg)n or (Lys)n polypeptides). Cells will be treated with a range of oligomer concentrations between 10 and 1,000 nM. After 24 to 48 hours we will test the effectiveness of antisense blockers by (i) quantifying the level of mRNA containing the expanded CGG repeats by qRT-PCR; (ii) monitoring of lamina disorganization and formation of ubiquitin-positive nuclear inclusions by immunofluorescence; (iii) production of FMRpolyG by flow cytometry, luciferase assay and western blot as well as formation of nuclear aggregates; (iv) quantifying the level of FMRP protein synthesis by western blot. Finally, the antisense blockers that prove to be effective in inhibition of RAN translation in cellule and that not affect significant decrease of FMRP levels will be next tested in vivo in mice expressing expanded CGG repeats (see 2.2.3).
2.2.3. Objective 6: Application of predesigned small compounds which bind CGGexp and inhibit RAN translation from 5’UTR of FMR1 (the project for second PhD student)
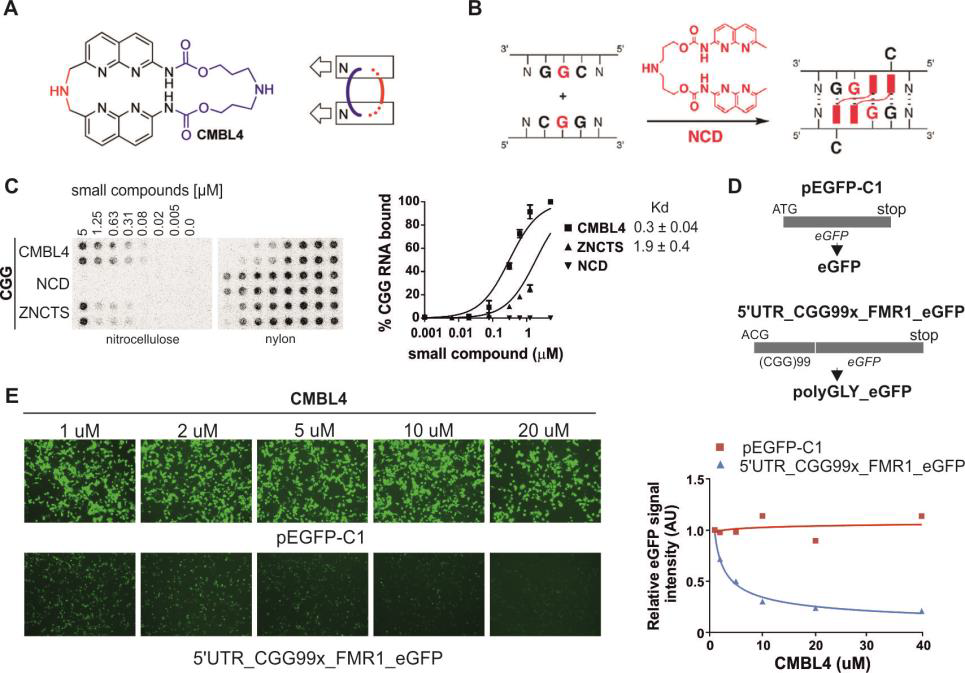
In parallel to antisense strategy, we will develop a CGG repeat-targeting small compounds strategy. As a proof of feasibility, more recently we screened small library of CMBL4 compounds, obtained from Dr. Kazuhiko Nakatani (Fig. 6a,b). They were design to bind two 5’-GG-3’ dinucleotide sequences located in front of each other on two interacting RNA strands. We noted, that they indeed bind specifically with nanomolar affinity to CGG repeat RNA hairpin in vitro (Fig. 6c). Moreover, treatment of engineered COS-7 cells with low micromolar CMBL4 induced significant reduction of FMRpolyG protein production and ineffective formation of nuclear aggregates (Fig. 6d,e). On the other hand the toxicity of this compound is relatively low (Fig. 6e, red line). Using neural cells obtained from inducible FXTAS mouse model the effect of the treatment was the same. Cortical neurons grown over three weeks in medium containing 500 nM CMBL4 the FMRpolyG aggregates were not detectable compare to control experiment.
In this project we are going to test new set of compounds designed, synthetized and characterized in Nakatani Lab at Osaka University (partner in this project). First, we will test the CGG RNA binding properties of compounds to calculate dissociation constant (Fig. 6c). Then, we will study the positive hits from in vitro screening by using simple COS-7 cell model and primary cortical or hippocampal neurons expressing expanded CGG repeats (described above). Briefly, we will test if our positive molecules reduce production of toxic FMRpolyG by western blot and flow cytometry, formation of nuclear aggregates by immunofluorescence using anti-FMRpolyG and ubiquitin-specific antibodies. In addition, we will test whether the identified compounds can restore normal DGCR8 localization by immunofluorescence, and normal SAM68-dependent alternative splicing by RT-PCR assays. Also, we will test the specificity of our positive molecules toward CGG RNA aggregates using other repeated sequences, such as expanded CAG, CUG and CCUG repeats, causing HD, DM1 and DM2 diseases, respectively. After first round of screening, the most potent molecules will be further modified by chemist from Osaka University and again tested using the battery of in vitro and in cellule assays. For selected compounds we want to study potential off-target effect on other RNAs in cells using RNA sequencing or microarray hybridization (Illumina). In the same time we will test the effect of compounds on expression of endogenous FMR1 (mRNA and protein level) in fibroblasts derived from FXTAS patients. The most promising hits identified in both small compounds screening and antisense blockers of RAN translation (2.2.3) will be evaluated in FXTAS inducible mouse models with a mutant (CGG)99 or normal (CGG)11 repeat containing RNA under control of a doxycycline responsive promoter (TetO-CGG99-EGFP and TetO-CGG11-EGFP) The expression of transgene RNA can be activated by adding doxycycline (dox) in the drinking water and subsequently inactivated following dox withdrawal. These mice already develop a significant number of nuclear aggregates in some neural cells and behavioral abnormalities upon 8 or 12 weeks of dox treatment. Administration of compounds and blockers will be performed using previously optimized local delivery system by ICV infusion with Alzet mini osmotic pumps. The administration of compounds and antisense blockers will be performed in Stork lab having longstanding experience in drug testing (partner in this project). Therapeutic efficacy will be established by measuring the Fmr1 mRNA levels (qRT-PCR), the number and/or size of nuclear RNA and FMRpolyG aggregates (FISH and immunostaining for ubiquitin), microRNA level by qRT-PCR and functional rescue (behavioral and physiological assessments). Evidence of possible reversibility of disease would be indicated by a reduction in neurological deficits in treated animals compared to their pre-treatment baseline performance. They already optimized all these assays [65].
References
[1] J.D. Cleary, L.P. Ranum, Repeat-associated non-ATG (RAN) translation in neurological disease, Hum Mol Genet 22(R1) (2013) R45-51.
[2] P.S. Harper, B.G. van Engelen, B. Eymard, M. Rogers, D. Wilcox, 99th ENMC international workshop: myotonic dystrophy: present management, future therapy. 9-11 November 2001, Naarden, The Netherlands, Neuromuscul Disord 12(6) (2002) 596-9.
[3] J.R. O’Rourke, M.S. Swanson, Mechanisms of RNA-mediated disease, J Biol Chem 284(12) (2009) 7419-23.
[4] X. Lin, J.W. Miller, A. Mankodi, R.N. Kanadia, Y. Yuan, R.T. Moxley, M.S. Swanson, C.A. Thornton, Failure of MBNL1-dependent postnatal splicing transitions in myotonic dystrophy, Hum Mol Genet 15(13) (2006) 2087-97.
[5] A. Mankodi, E. Logigian, L. Callahan, C. McClain, R. White, D. Henderson, M. Krym, C.A. Thornton, Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat, Science 289(5485) (2000) 1769-73.
[6] R.N. Kanadia, K.A. Johnstone, A. Mankodi, C. Lungu, C.A. Thornton, D. Esson, A.M. Timmers, W.W. Hauswirth, M.S. Swanson, A muscleblind knockout model for myotonic dystrophy, Science 302(5652) (2003) 1978-80.
[7] J.P. Orengo, P. Chambon, D. Metzger, D.R. Mosier, G.J. Snipes, T.A. Cooper, Expanded CTG repeats within the DMPK 3′ UTR causes severe skeletal muscle wasting in an inducible mouse model for myotonic dystrophy, Proc Natl Acad Sci U S A 105(7) (2008) 2646-51.
[8] R. Batra, D.A. Nelles, E. Pirie, S.M. Blue, R.J. Marina, H. Wang, I.A. Chaim, J.D. Thomas, N. Zhang, V. Nguyen, S. Aigner, S. Markmiller, G. Xia, K.D. Corbett, M.S. Swanson, G.W. Yeo, Elimination of Toxic Microsatellite Repeat Expansion RNA by RNA-Targeting Cas9, Cell 170(5) (2017) 899-912.e10.
[9] K. Sobczak, T.M. Wheeler, W. Wang, C.A. Thornton, RNA interference targeting CUG repeats in a mouse model of myotonic dystrophy, Mol Ther 21(2) (2013) 380-7.
[10] J.E. Lee, C.F. Bennett, T.A. Cooper, RNase H-mediated degradation of toxic RNA in myotonic dystrophy type 1, Proc Natl Acad Sci U S A 109(11) (2012) 4221-6.
[11] T.M. Wheeler, A.J. Leger, S.K. Pandey, A.R. MacLeod, M. Nakamori, S.H. Cheng, B.M. Wentworth, C.F. Bennett, C.A. Thornton, Targeting nuclear RNA for in vivo correction of myotonic dystrophy, Nature 488(7409) (2012) 111-5.
[12] A. Wojtkowiak-Szlachcic, K. Taylor, E. Stepniak-Konieczna, L.J. Sznajder, A. Mykowska, J. Sroka, C.A. Thornton, K. Sobczak, Short antisense-locked nucleic acids (all-LNAs) correct alternative splicing abnormalities in myotonic dystrophy, Nucleic Acids Res 43(6) (2015) 3318-31.
[13] T.M. Wheeler, K. Sobczak, J.D. Lueck, R.J. Osborne, X. Lin, R.T. Dirksen, C.A. Thornton, Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA, Science 325(5938) (2009) 336-9.
[14] P. Konieczny, E. Stepniak-Konieczna, K. Sobczak, MBNL proteins and their target RNAs, interaction and splicing regulation, Nucleic Acids Res 42(17) (2014) 10873-87.
[15] W.J. Krzyzosiak, K. Sobczak, M. Wojciechowska, A. Fiszer, A. Mykowska, P. Kozlowski, Triplet repeat RNA structure and its role as pathogenic agent and therapeutic target, Nucleic Acids Res 40(1) (2012) 11-26.
[16] R.N. Kanadia, J. Shin, Y. Yuan, S.G. Beattie, T.M. Wheeler, C.A. Thornton, M.S. Swanson, Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy, Proc Natl Acad Sci U S A 103(31) (2006) 11748-53.
[17] L.J. Sznajder, M. Michalak, K. Taylor, P. Cywoniuk, M. Kabza, A. Wojtkowiak-Szlachcic, M. Matloka, P. Konieczny, K. Sobczak, Mechanistic determinants of MBNL activity, Nucleic Acids Res 44(21) (2016) 10326-10342.
[18] H. Du, M.S. Cline, R.J. Osborne, D.L. Tuttle, T.A. Clark, J.P. Donohue, M.P. Hall, L. Shiue, M.S. Swanson, C.A. Thornton, M. Ares, Jr., Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy, Nat Struct Mol Biol 17(2) (2010) 187-93.
[19] E.T. Wang, N.A. Cody, S. Jog, M. Biancolella, T.T. Wang, D.J. Treacy, S. Luo, G.P. Schroth, D.E. Housman, S. Reddy, E. Lecuyer, C.B. Burge, Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins, Cell 150(4) (2012) 710- 24.
[20] R. Batra, M. Manchanda, M.S. Swanson, Global insights into alternative polyadenylation regulation, RNA Biol 12(6) (2015) 597-602.
[21] Y. Adereth, V. Dammai, N. Kose, R. Li, T. Hsu, RNA-dependent integrin alpha3 protein localization regulated by the Muscleblind-like protein MLP1, Nat Cell Biol 7(12) (2005) 1240-7.
[22] A. Masuda, H.S. Andersen, T.K. Doktor, T. Okamoto, M. Ito, B.S. Andresen, K. Ohno, CUGBP1 and MBNL1 preferentially bind to 3′ UTRs and facilitate mRNA decay, Sci Rep 2 (2012) 209.
[23] F. Rau, F. Freyermuth, C. Fugier, J.P. Villemin, M.C. Fischer, B. Jost, D. Dembele, G. Gourdon, A. Nicole, D. Duboc, K. Wahbi, J.W. Day, H. Fujimura, M.P. Takahashi, D. Auboeuf, N. Dreumont, D. Furling, N. Charlet-Berguerand, Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy, Nat Struct Mol Biol 18(7) (2011) 840-5.
[24] K. Charizanis, K.Y. Lee, R. Batra, M. Goodwin, C. Zhang, Y. Yuan, L. Shiue, M. Cline, M.M. Scotti, G. Xia, A. Kumar, T. Ashizawa, H.B. Clark, T. Kimura, M.P. Takahashi, H. Fujimura, K. Jinnai, H. Yoshikawa, M. Gomes-Pereira, G. Gourdon, N. Sakai, S. Nishino, T.C. Foster, M. Ares, Jr., R.B. Darnell, M.S. Swanson, Muscleblind-like 2-mediated alternative splicing in the developing brain and
dysregulation in myotonic dystrophy, Neuron 75(3) (2012) 437-50.
[25] I. Holt, V. Jacquemin, M. Fardaei, C.A. Sewry, G.S. Butler-Browne, D. Furling, J.D. Brook, G.E. Morris, Muscleblind-like proteins: similarities and differences in normal and myotonic dystrophy muscle, Am J Pathol 174(1) (2009) 216-27.
[26] K.S. Lee, R.M. Squillace, E.H. Wang, Expression pattern of muscleblind-like proteins differs in differentiating myoblasts, Biochem Biophys Res Commun 361(1) (2007) 151-5.
[27] M. Fardaei, M.T. Rogers, H.M. Thorpe, K. Larkin, M.G. Hamshere, P.S. Harper, J.D. Brook, Three proteins, MBNL, MBLL and MBXL, colocalize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells, Hum Mol Genet 11(7) (2002) 805-14.
[28] M. Teplova, D.J. Patel, Structural insights into RNA recognition by the alternative-splicing regulator muscleblind-like MBNL1, Nat Struct Mol Biol 15(12) (2008) 1343-51.
[29] D. Cass, R. Hotchko, P. Barber, K. Jones, D.P. Gates, J.A. Berglund, The four Zn fingers of MBNL1 provide a flexible platform for recognition of its RNA binding elements, BMC Mol Biol 12 (2011) 20.
[30] S. Park, P.D. Phukan, M. Zeeb, M.A. Martinez-Yamout, H.J. Dyson, P.E. Wright, Structural Basis for Interaction of the Tandem Zinc Finger Domains of Human Muscleblind with Cognate RNA from Human Cardiac Troponin T, Biochemistry 56(32) (2017) 4154-4168.
[31] I. Grammatikakis, Y.H. Goo, G.V. Echeverria, T.A. Cooper, Identification of MBNL1 and MBNL3 domains required for splicing activation and repression, Nucleic Acids Res 39(7) (2011) 2769-80.
[32] H. Tran, N. Gourrier, C. Lemercier-Neuillet, C.M. Dhaenens, A. Vautrin, F.J. Fernandez-Gomez, L. Arandel, C. Carpentier, H. Obriot, S. Eddarkaoui, L. Delattre, E. Van Brussels, I. Holt, G.E. Morris, B. Sablonniere, L. Buee, N. Charlet-Berguerand, S. Schraen-Maschke, D. Furling, I. Behm-Ansmant, C. Branlant, M.L. Caillet-Boudin, N. Sergeant, Analysis of exonic regions involved in nuclear localization, splicing activity, and dimerization of Muscleblind-like-1 isoforms, J Biol Chem 286(18) (2011) 16435-46.
[33] C. Edge, C. Gooding, C.W. Smith, Dissecting domains necessary for activation and repression of splicing by Muscleblind-like protein 1, BMC Mol Biol 14 (2013) 29.
[34] M.G. Poulos, R. Batra, M. Li, Y. Yuan, C. Zhang, R.B. Darnell, M.S. Swanson, Progressive impairment of muscle regeneration in muscleblind-like 3 isoform knockout mice, Hum Mol Genet 22(17) (2013) 3547-58.
[35] H. Tran, N. Gourrier, C. Lemercier-Neuillet, C.M. Dhaenens, A. Vautrin, F.J. Fernandez-Gomez, L. Arandel, C. Carpentier, H. Obriot, S. Eddarkaoui, L. Delattre, E. Van Brussels, I. Holt, G.E. Morris, B. Sablonnière, L. Buée, N. Charlet-Berguerand, S. Schraen-Maschke, D. Furling, I. Behm-Ansmant, C. Branlant, M.L. Caillet-Boudin, N. Sergeant, Analysis of exonic regions involved in nuclear localization, splicing activity, and dimerization of Muscleblind-like-1 isoforms, J Biol Chem 286(18) (2011) 16435-46.
[36] F. Terenzi, A.N. Ladd, Conserved developmental alternative splicing of muscleblind-like (MBNL) transcripts regulates MBNL localization and activity, RNA Biol 7(1) (2010) 43-55.
[37] Y. Yuan, S.A. Compton, K. Sobczak, M.G. Stenberg, C.A. Thornton, J.D. Griffith, M.S. Swanson, Muscleblind-like 1 interacts with RNA hairpins in splicing target and pathogenic RNAs, Nucleic Acids Res 35(16) (2007) 5474-86.
[38] Y. Fu, S.R. Ramisetty, N. Hussain, A.M. Baranger, MBNL1-RNA recognition: contributions of MBNL1 sequence and RNA conformation, Chembiochem 13(1) (2012) 112-9.
[39] N. Lambert, A. Robertson, M. Jangi, S. McGeary, P.A. Sharp, C.B. Burge, RNA Bind-n-Seq: quantitative assessment of the sequence and structural binding specificity of RNA binding proteins, Mol Cell 54(5) (2014) 887-900.
[40] G.V. Echeverria, T.A. Cooper, Muscleblind-like 1 activates insulin receptor exon 11 inclusion by enhancing U2AF65 binding and splicing of the upstream intron, Nucleic Acids Res 42(3) (2014) 1893-903.
[41] M.B. Warf, J.V. Diegel, P.H. von Hippel, J.A. Berglund, The protein factors MBNL1 and U2AF65 bind alternative RNA structures to regulate splicing, Proc Natl Acad Sci U S A 106(23) (2009) 9203-8.
[42] E.T. Wang, A.J. Ward, J.M. Cherone, J. Giudice, T.T. Wang, D.J. Treacy, N.J. Lambert, P. Freese, T. Saxena, T.A. Cooper, C.B. Burge, Antagonistic regulation of mRNA expression and splicing by CELF and MBNL proteins, Genome Res 25(6) (2015) 858-71.
[43] R. Klinck, A. Fourrier, P. Thibault, J. Toutant, M. Durand, E. Lapointe, M.L. Caillet-Boudin, N. Sergeant, G. Gourdon, G. Meola, D. Furling, J. Puymirat, B. Chabot, RBFOX1 cooperates with MBNL1 to control splicing in muscle, including events altered in myotonic dystrophy type 1, PLoS One 9(9) (2014) e107324.
[44] F.X. Laurent, A. Sureau, A.F. Klein, F. Trouslard, E. Gasnier, D. Furling, J. Marie, New function for the RNA helicase p68/DDX5 as a modifier of MBNL1 activity on expanded CUG repeats, Nucleic Acids Res 40(7) (2012) 3159-71.
[45] P. Cywoniuk, K. Taylor, Ł. Sznajder, K. Sobczak, Hybrid splicing minigene and antisense oligonucleotides as efficient tools to determine functional protein/RNA interactions, Sci Rep 7(1) (2017) 17587.
[46] R.J. Hagerman, M. Leehey, W. Heinrichs, F. Tassone, R. Wilson, J. Hills, J. Grigsby, B. Gage, P.J. Hagerman, Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X, Neurology 57(1) (2001) 127-30.
[47] F. Tassone, K.P. Iong, T.H. Tong, J. Lo, L.W. Gane, E. Berry-Kravis, D. Nguyen, L.Y. Mu, J. Laffin, D.B. Bailey, R.J. Hagerman, FMR1 CGG allele size and prevalence ascertained through newborn screening in the United States, Genome Med 4(12) (2012) 100.
[48] S. Jacquemont, R.J. Hagerman, M. Leehey, J. Grigsby, L. Zhang, J.A. Brunberg, C. Greco, V. Des Portes, T. Jardini, R. Levine, E. Berry- Kravis, W.T. Brown, S. Schaeffer, J. Kissel, F. Tassone, P.J. Hagerman, Fragile X premutation tremor/ataxia syndrome: molecular, clinical, and neuroimaging correlates, Am J Hum Genet 72(4) (2003) 869-78.
[49] C.M. Greco, R.F. Berman, R.M. Martin, F. Tassone, P.H. Schwartz, A. Chang, B.D. Trapp, C. Iwahashi, J. Brunberg, J. Grigsby, D. Hessl, E.J. Becker, J. Papazian, M.A. Leehey, R.J. Hagerman, P.J. Hagerman, Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS), Brain 129(Pt 1) (2006) 243-55.
[50] F. Tassone, R.J. Hagerman, D.Z. Loesch, A. Lachiewicz, A.K. Taylor, P.J. Hagerman, Fragile X males with unmethylated, full mutation trinucleotide repeat expansions have elevated levels of FMR1 messenger RNA, Am J Med Genet 94(3) (2000) 232-6.
[51] R. Willemsen, J. Levenga, B.A. Oostra, CGG repeat in the FMR1 gene: size matters, Clin Genet 80(3) (2011) 214-25.
[52] C. Sellier, F. Freyermuth, R. Tabet, T. Tran, F. He, F. Ruffenach, V. Alunni, H. Moine, C. Thibault, A. Page, F. Tassone, R. Willemsen, M.D. Disney, P.J. Hagerman, P.K. Todd, N. Charlet-Berguerand, Sequestration of DROSHA and DGCR8 by expanded CGG RNA repeats alters microRNA processing in fragile X-associated tremor/ataxia syndrome, Cell Rep 3(3) (2013) 869-80.
[53] C. Sellier, F. Rau, Y. Liu, F. Tassone, R.K. Hukema, R. Gattoni, A. Schneider, S. Richard, R. Willemsen, D.J. Elliott, P.J. Hagerman, N. Charlet-Berguerand, Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients, EMBO J 29(7) (2010) 1248-61.
[54] P.K. Todd, S.Y. Oh, A. Krans, F. He, C. Sellier, M. Frazer, A.J. Renoux, K.C. Chen, K.M. Scaglione, V. Basrur, K. Elenitoba-Johnson, J.P. Vonsattel, E.D. Louis, M.A. Sutton, J.P. Taylor, R.E. Mills, N. Charlet-Berguerand, H.L. Paulson, CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome, Neuron 78(3) (2013) 440-55.
[55] M.G. Kearse, K.M. Green, A. Krans, C.M. Rodriguez, A.E. Linsalata, A.C. Goldstrohm, P.K. Todd, CGG Repeat-Associated Non-AUG Translation Utilizes a Cap-Dependent Scanning Mechanism of Initiation to Produce Toxic Proteins, Mol Cell 62(2) (2016) 314-22.
[56] C. Sellier, R.A.M. Buijsen, F. He, S. Natla, L. Jung, P. Tropel, A. Gaucherot, H. Jacobs, H. Meziane, A. Vincent, M.F. Champy, T. Sorg, G. Pavlovic, M. Wattenhofer-Donze, M.C. Birling, M. Oulad-Abdelghani, P. Eberling, F. Ruffenach, M. Joint, M. Anheim, V. Martinez- Cerdeno, F. Tassone, R. Willemsen, R.K. Hukema, S. Viville, C. Martinat, P.K. Todd, N. Charlet-Berguerand, Translation of Expanded CGG Repeats into FMRpolyG Is Pathogenic and May Contribute to Fragile X Tremor Ataxia Syndrome, Neuron 93(2) (2017) 331-347.
[57] R.A. Buijsen, C. Sellier, L.A. Severijnen, M. Oulad-Abdelghani, R.F. Verhagen, R.F. Berman, N. Charlet-Berguerand, R. Willemsen, R.K. Hukema, FMRpolyG-positive inclusions in CNS and non-CNS organs of a fragile X premutation carrier with fragile X-associated tremor/ataxia syndrome, Acta Neuropathol Commun 2 (2014) 162.
[58] M. Boivin, R. Willemsen, R.K. Hukema, C. Sellier, Potential pathogenic mechanisms underlying Fragile X Tremor Ataxia Syndrome: RAN translation and/or RNA gain-of-function?, Eur J Med Genet (2017).
[59] F. He, A. Krans, B.D. Freibaum, J.P. Taylor, P.K. Todd, TDP-43 suppresses CGG repeat-induced neurotoxicity through interactions with HnRNP A2/B1, Hum Mol Genet 23(19) (2014) 5036-51.
[60] R.F. Berman, R.A. Buijsen, K. Usdin, E. Pintado, F. Kooy, D. Pretto, I.N. Pessah, D.L. Nelson, Z. Zalewski, N. Charlet-Bergeurand, R. Willemsen, R.K. Hukema, Mouse models of the fragile X premutation and fragile X-associated tremor/ataxia syndrome, J Neurodev Disord 6(1) (2014) 25.
[61] M.M. Foote, M. Careaga, R.F. Berman, What has been learned from mouse models of the Fragile X Premutation and Fragile Xassociated tremor/ataxia syndrome?, Clin Neuropsychol 30(6) (2016) 960-72.
[62] R. Willemsen, M. Hoogeveen-Westerveld, S. Reis, J. Holstege, L.A. Severijnen, I.M. Nieuwenhuizen, M. Schrier, L. van Unen, F. Tassone, A.T. Hoogeveen, P.J. Hagerman, E.J. Mientjes, B.A. Oostra, The FMR1 CGG repeat mouse displays ubiquitin-positive intranuclear neuronal inclusions; implications for the cerebellar tremor/ataxia syndrome, Hum Mol Genet 12(9) (2003) 949-59.
[63] A. Entezam, R. Biacsi, B. Orrison, T. Saha, G.E. Hoffman, E. Grabczyk, R.L. Nussbaum, K. Usdin, Regional FMRP deficits and large repeat expansions into the full mutation range in a new Fragile X premutation mouse model, Gene 395(1-2) (2007) 125-34.
[64] R.K. Hukema, R.A. Buijsen, M. Schonewille, C. Raske, L.A. Severijnen, I. Nieuwenhuizen-Bakker, R.F. Verhagen, L. van Dessel, A. Maas, N. Charlet-Berguerand, C.I. De Zeeuw, P.J. Hagerman, R.F. Berman, R. Willemsen, Reversibility of neuropathology and motor deficits in an inducible mouse model for FXTAS, Hum Mol Genet 24(17) (2015) 4948-57.
[65] H. Castro, E. Kul, R.A.M. Buijsen, L.W.F.M. Severijnen, R. Willemsen, R.K. Hukema, O. Stork, M. Santos, Selective rescue of heightened anxiety but not gait ataxia in a premutation 90CGG mouse model of Fragile X-associated tremor/ataxia syndrome, Hum Mol Genet 26(11) (2017) 2133-2145.
[66] W.Y. Yang, H.D. Wilson, S.P. Velagapudi, M.D. Disney, Inhibition of Non-ATG Translational Events in Cells via Covalent Small Molecules Targeting RNA, J Am Chem Soc 137(16) (2015) 5336-45.
[67] M.D. Disney, B. Liu, W.Y. Yang, C. Sellier, T. Tran, N. Charlet-Berguerand, J.L. Childs-Disney, A small molecule that targets r(CGG)(exp) and improves defects in fragile X-associated tremor ataxia syndrome, ACS Chem Biol 7(10) (2012) 1711-8.
[68] T. Tran, J.L. Childs-Disney, B. Liu, L. Guan, S. Rzuczek, M.D. Disney, Targeting the r(CGG) repeats that cause FXTAS with modularly assembled small molecules and oligonucleotides, ACS Chem Biol 9(4) (2014) 904-12.
[69] K. Zawadzka, P. Zawadzki, R. Baker, K.V. Rajasekar, F. Wagner, D.J. Sherratt, L.K. Arciszewska, MukB ATPases are regulated independently by the N- and C-terminal domains of MukF kleisin, Elife 7 (2018).
[70] D. Duchi, D.L. Bauer, L. Fernandez, G. Evans, N. Robb, L.C. Hwang, K. Gryte, A. Tomescu, P. Zawadzki, Z. Morichaud, K. Brodolin, A.N. Kapanidis, RNA Polymerase Pausing during Initial Transcription, Mol Cell 63(6) (2016) 939-50.
[71] M. Stracy, M. Jaciuk, S. Uphoff, A.N. Kapanidis, M. Nowotny, D.J. Sherratt, P. Zawadzki, Single-molecule imaging of UvrA and UvrB recruitment to DNA lesions in living Escherichia coli, Nat Commun 7 (2016) 12568.
[72] P.F. May, P. Zawadzki, D.J. Sherratt, A.N. Kapanidis, L.K. Arciszewska, Assembly, translocation, and activation of XerCD-dif recombination by FtsK translocase analyzed in real-time by FRET and two-color tethered fluorophore motion, Proc Natl Acad Sci U S A 112(37) (2015) E5133-41.
[73] M. Stracy, C. Lesterlin, F. Garza de Leon, S. Uphoff, P. Zawadzki, A.N. Kapanidis, Live -cell superresolu_on microscopy reveals the organiza_on of RNA polymerase in the bacterial nucleoid, Proc Natl Acad Sci U S A 112(32) (2015) E4390 -9.
[74] P. Zawadzki, M. Stracy, K. Ginda, K. Zawadzka, C. Lesterlin, A.N. Kapanidis, D.J. Sherra_, The Localiza_on and Ac_on o f Topoisomerase IV in Escherichia coli Chromosome Segrega_on Is Coordinated by the SMC Complex, MukBEF, Cell Rep 13(11) (2015) 2587-2596.
[75] P.F.J. May, J.N.M. Pinkney, P. Zawadzki, G.W. Evans, D.J. Sherra_, A.N. Kapanidis, Tethered fluorophore mo_on: studying large DNA conforma_onal changes by single-fluorophore imaging, Biophys J 107(5) (2014) 1205-1216.
[76] P. Zawadzki, P.F. May, R.A. Baker, J.N. Pinkney, A.N. Kapanidis, D.J. Sherra_, L.K. Arciszewska, Conforma_onal transi_ons during FtsK translocase ac_va_on of individual XerCD-dif recombina_on complexes, Proc Natl Acad Sci U S A 110(43) (2013) 17302-7.
[77] J.N. Pinkney, P. Zawadzki, J. Mazuryk, L.K. Arciszewska, D.J. Sherra_, A.N. Kapanidis, Capturing reac_on paths and intermediates in Cre-loxP recombina_on using single-molecule fluorescence, Proc Natl Acad Sci U S A 109(51) (2012) 20871-6.
[78] E. Allemand, M.P. Myers, J. Garcia -Bernardo, A. Harel-Bellan, A.R. Krainer, C. Muchardt, A Broad Set of Chroma_n Factors Influences Splicing, PLoS Genet 12(9) (2016) e1006318.
[79] M. Weber, D. Schneider, Six amino acids define a minimal dimeriza_on sequence and stabilize a transmembrane helix dimer by close packing and hydrogen bonding, FEBS Le_ 587(11) (2013) 1592-6.
[80] P. Konieczny, E. Stepniak -Konieczna, K. Taylor, L.J. Sznajder, K. Sobczak, Autoregula_on of MBNL1 func_on by exon 1 exclusion from MBNL1 transcript, Nucleic Acids Res 45(4) (2017) 1760-1775.
[81] R. Rovozzo, G. Korza, M.W. Baker, M. Li, A. Bha_acharyya, E. Barbarese, J.H. Carson, CGG Repeats in the 5’UTR of FMR1 RN A Regulate Transla_on of Other RNAs Localized in the Same RNA Granules, PLoS One 11(12) (2016) e0168204.
[82] K. Sobczak, G. Michlewski, M. de Mezer, J. Krol, W.J. Krzyzosiak, Trinucleo_de repeat system for sequence specificity an alysis of RNA structure probing reagents, Anal Biochem 402(1) (2010) 40-6.
[83] K. Sobczak, M. de Mezer, G. Michlewski, J. Krol, W.J. Krzyzosiak, RNA structure of trinucleo_de repeats associated with human neurological diseases, Nucleic Acids Res 31(19) (2003) 5469-82.
[84] J.K. Davis, K. Broadie, Mul_farious Func_ons of the Fragile X Mental R etarda_on Protein, Trends Genet 33(10) (2017) 703-714.
[85] E. Fernández, N. Rajan, C. Bagni, The FMRP regulon: from targets to disease convergence, Front Neurosci 7 (2013) 191.
[86] M.C. Didiot, Z. Tian, C. Schaeffer, M. Subramanian, J.L. Mandel, H. Moine, The G-quartet containing FMRP binding site in FMR1 mRNA is a potent exonic splicing enhancer, Nucleic Acids Res 36(15) (2008) 4902 -12.
[87] A.C. Blice-Baum, M.R. Mihailescu, Biophysical characteriza_on of G-quadruplex forming FMR1 mRNA and of its interac_ons with different fragile X mental retarda_on protein isoforms, RNA 20(1) (2014) 103 -14.
[88] C. Schaeffer, B. Bardoni, J.L. Mandel, B. Ehresmann, C. Ehresmann, H. Moine, The fragile X mental retarda_on protein bin ds specifically to its mRNA via a purine quartet mo_f, EMBO J 20(17) (2001) 4803-13.
[89] A. Cammas, S. Millevoi, RNA G-quadruplexes: emerging mechanisms in disease, Nucleic Acids Res 45(4) (2017) 1584-1595.
[90] K. Sobczak, G. Michlewski, M. de Mezer, E. Kierzek, J. Krol, M. O lejniczak, R. Kierzek, W.J. Krzyzosiak, Structural diversity of triplet repeat RNAs, J Biol Chem 285(17) (2010) 12755-64.
[91] K. Lepeta, K.J. Purzycka, K. Pachulska-Wieczorek, M. Mitjans, M. Begemann, B. Vafadari, K. Bijata, R.W. Adamiak, H. Ehrenreich, M. Dziembowska, L. Kaczmarek, A normal gene_c varia_on modulates synap_c MMP -9 protein levels and the severity of schizophrenia symptoms, EMBO Mol Med 9(8) (2017) 1100-1116.
[92] S. Mukherjee, C. Dohno, K. Nakatani, Design and Synthesis of Cyclic Mismatch-Binding Ligands (CMBLs) with Variable Linkers by Ring- Closing Metathesis and their Photophysical and DNA Repeat Binding Proper_es, Chemistry 23(47) (2017) 11385 -11396.
[93] C. Dohno, I. Kohyama, C. Hong, K. Nakatani, Naphthyridine tetramer with a pre-organized structure for 1:1 binding to a CGG/CGG sequence, Nucleic Acids Res 40(6) (2012) 2771-81.
[94] C. Dohno, H. Atsumi, K. Nakatani, Ligand inducible assembly of a DNA tetrahedron, Chem Commun (Camb) 47(12) (2011) 3499-501.